
Abstract
This report presents a rare case of a 4.5-year-old boy with β-thalassemia major, who developed acute myeloid leukemia (AML) French-American-British type M2. β-thalassemia major is an inherited hemoglobinopathy characterized by severe, transfusion-dependent anemia. Chronic blood transfusions, while lifesaving, can lead to iron overload, increasing the risk of secondary malignancies like leukemia due to free radical generation. Our case emphasizes the need for careful monitoring of thalassemia patients for secondary malignancies. The patient, born to consanguineous parents, presented with symptoms such as persistent fever, abdominal pain, and splenomegaly. Hematological investigations revealed severe cytopenias with a high blast count. Bone marrow examination confirmed AML M2. Despite aggressive chemotherapy, the patient sadly passed away within a month of diagnosis.
introduction
β-thalassemia encompasses a group of inherited disorders caused by decreased or absent β-globin chain synthesis, leading to α-globin chain accumulation in red blood cells (RBCs). It is an autosomal recessive disorder, with β-thalassemia major being its most severe form. Lifelong blood transfusions are required for survival, but these transfusions carry the risk of iron overload, which can be harmful to various organs. Stem cell transplantation remains the only definitive cure, although supportive care, including iron chelation, is essential. Patients with β-thalassemia major are increasingly reported to have a higher risk of developing malignancies like leukemia at an early age, emphasizing the need for early cancer screening.
Case Presentation
History of Presentation
A 4.5-year-old boy, born to consanguineous parents, was diagnosed with thalassemia major at six months old. His transfusion needs increased with age, requiring weekly blood transfusions by four years. Despite the frequency of transfusions, there was no evaluation for iron overload or initiation of iron chelation therapy.
Physical Examination
The child presented with a two-week history of fever, abdominal pain, fatigue, and poor appetite. Examination revealed significant pallor, lethargy, bruising on the legs, and massive splenomegaly extending to the right iliac fossa. No hepatomegaly was observed.
Laboratory Findings
Laboratory tests indicated hypochromic, microcytic red cells with fragmented forms. Blasts were present in the blood smear, prompting further investigation with a bone marrow examination.
| Parameters | Patients values | Reference values |
|---|---|---|
| Complete blood count | ||
| Hemoglobin (g/dL) | 5.3 | 12-15 |
| Platelet count (×109/L) | 16 | 150-450 |
| Total leukocyte count (×109/L) | 33 | 4-19 |
| Differential count | ||
| Neutrophils (%) | 7 | 40-80 |
| Lymphocytes (%) | 37 | 20-40 |
| Monocytes (%) | 2 | 2-10 |
| Eosinophils (%) | 0 | 1-6 |
| Myelocytes (%) | 3 | 0 |
| Blast cells (%) | 51 | 0 |
| Nucleated RBCs | 130 nucleated RBCs/100 WBCs | 0 |
| Serum iron profile | ||
| Serum ferritin (ng/mL) | >4,000 | 30-350 |
Table 1: Laboratory findings
RBCs: red blood cells; WBCs: white blood cells
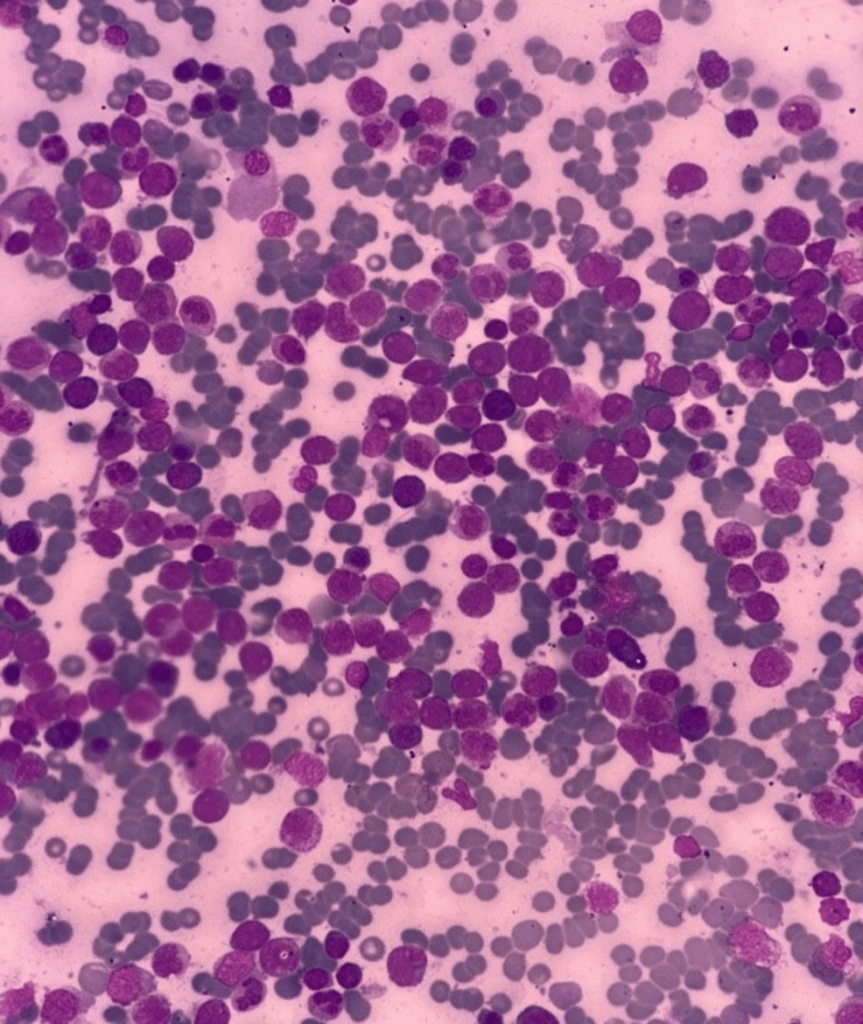
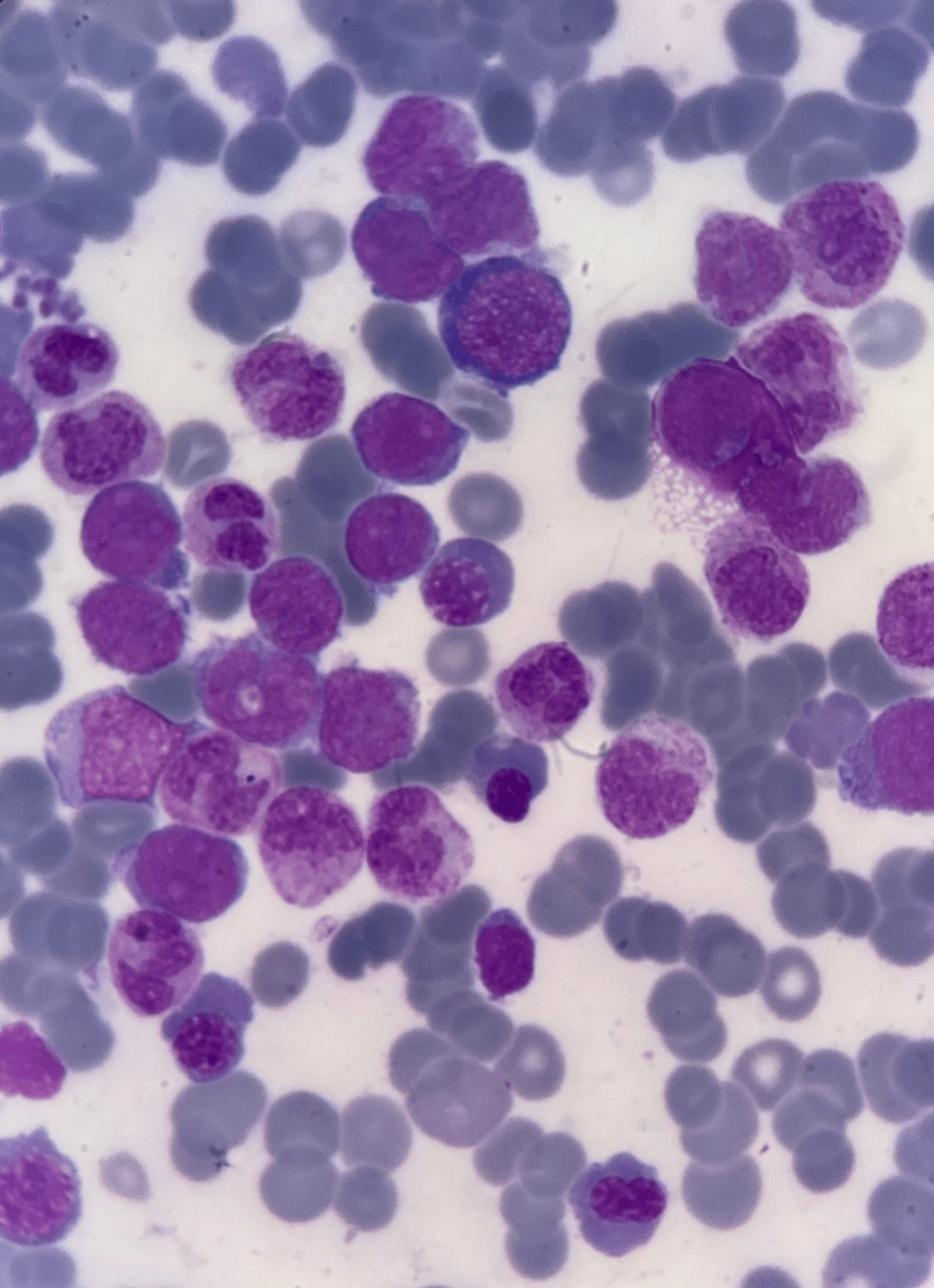
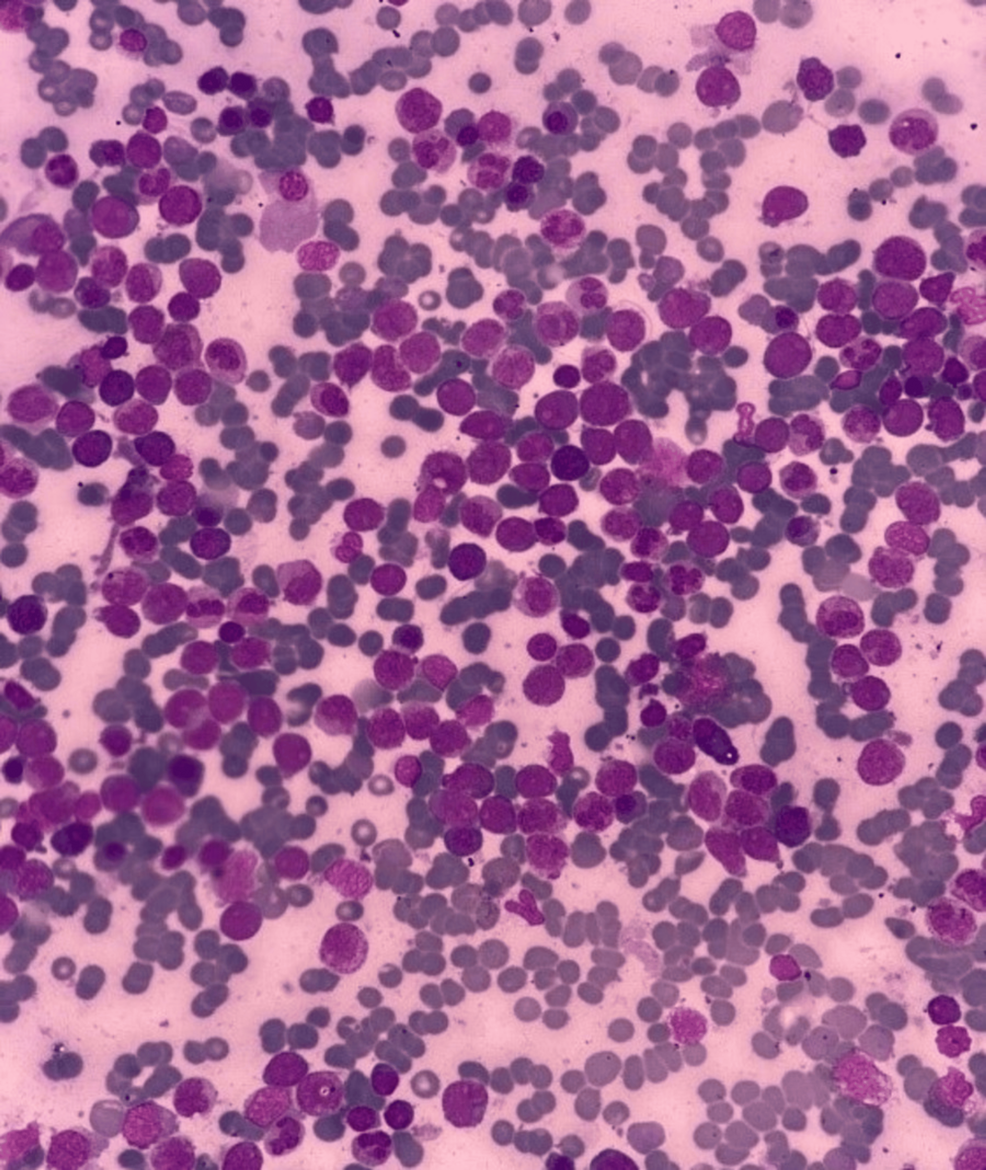
Bone Marrow Evaluation
Bone marrow aspiration revealed hypercellular fragments with normoblastic erythropoiesis and megaloblastic changes. Myelopoiesis was reduced, and 22% of the marrow consisted of blasts. A positive Sudan black B stain confirmed the myeloid origin of the blasts. Further immunophenotyping indicated AML M2.
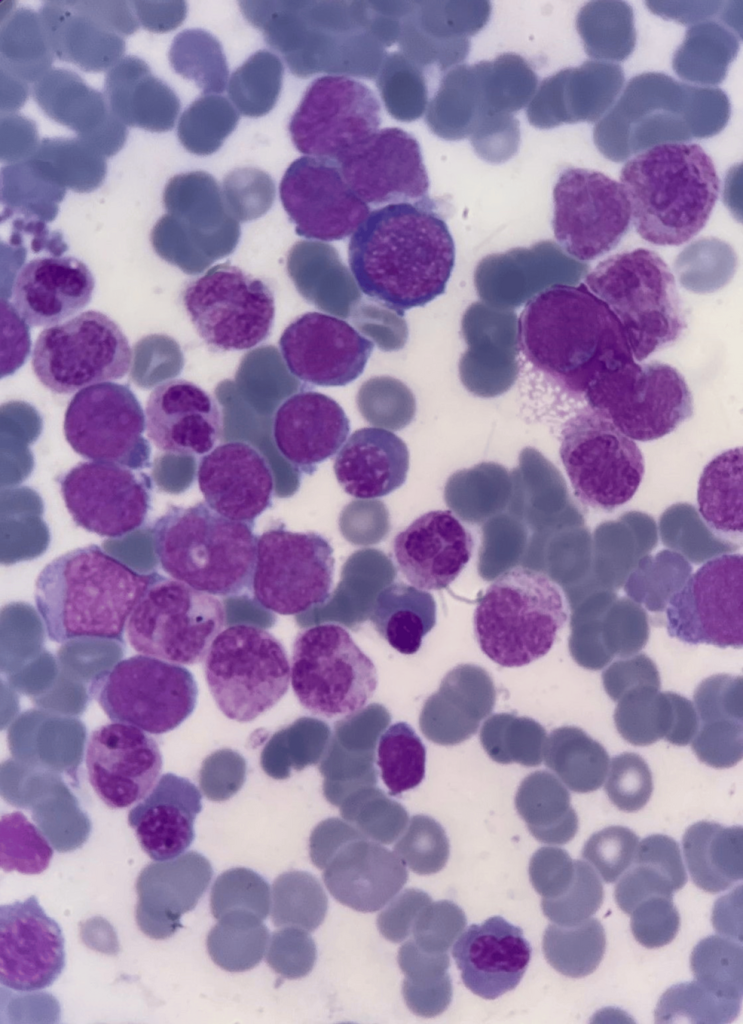
FAB: French-American-British

FAB: French-American-British
Diagnosis
A diagnosis of AML M2 was confirmed based on peripheral blood smear, bone marrow biopsy, and immunophenotyping. Polymerase chain reaction tests for AML markers were negative.
Treatment
Chemotherapy was initiated for AML M2, and the patient’s marrow blast count dropped below 2% by the eighth day of treatment. Despite an initial response, the child could not tolerate the treatment and died within one month.
Discussion
β-thalassemia is a genetic disorder caused by a deficiency in β-globin chain production. The resulting imbalance with α-globin chains leads to ineffective erythropoiesis and anemia. While treatments such as blood transfusions and iron chelation have improved life expectancy, iron overload remains a significant challenge, especially with repeated transfusions. Excess iron generates reactive oxygen species, leading to oxidative stress, which may increase the risk of malignancies. Although the exact link between iron overload and cancer development remains unclear, it is crucial to establish cancer screening protocols for thalassemia patients.
The development of malignancies like leukemia in β-thalassemia patients might be influenced by iron overload and transfusion-related immunomodulation. With improved life expectancy comes the emergence of these complications, necessitating a multidisciplinary approach for their management. Research into the connection between iron overload and cancer is vital to improve patient care outcomes.
Conclusions
β-thalassemia is a multifaceted genetic disorder requiring ongoing care and monitoring. Advances in treatment have improved survival, but iron overload presents new challenges, including the potential risk of malignancies. Regular cancer screenings and a multidisciplinary approach are essential to detect and manage secondary complications like AML in thalassemia patients. Further research is needed to understand the precise mechanisms of malignancy development in this population.
Thalassemia #AcuteMyeloidLeukemia #AML #PediatricCancer #BloodDisorders #IronOverload #CancerScreening #MedicalCaseStudy #Hematology

+ There are no comments
Add yours